Мы поможем в написании ваших работ!
ЗНАЕТЕ ЛИ ВЫ?
|
Наследственные (первичные) энзимопатии: нарушение обмена при алкаптонурии, фенилкетонурии, гипераммониемии.
Содержание книги
- Биологические функции белков. Взаимодействие с лигандами.
- Конформация белковых молекул (вторичная и третичная структуры). Активный центр белков. Связывание лигандов.
- Основы кинетики ферментативного катализа
- Кофакторы ферментов. Коферментные функции витаминов группы В.
- Ингибиторы активности ферментов
- Классификация и номенклатура ферментов
- Наследственные (первичные) энзимопатии: нарушение обмена при алкаптонурии, фенилкетонурии, гипераммониемии.
- Активаторы и ингибиторы ферментов
- Регуляция ферментов путем их фосфорилирования — дефосфорилирования
- Процесс включает следующие основные этапы.
- III. Исключение праймеров. Завершение формирования отстающей цепи ДНК
- Биосинтез рнк (транскрипция). Посттранскрипционные модификации рнк
- Биосинтез белков. Основные компоненты и этапы этого процесса. Посттрансляционный процессинг белков.
- Регуляция экспрессии генов. Теория оперона. Индукция и репрессия синтеза белков.
- Молекулярные механизмы генетической изменчивости. Мутации. Наследственные протеинопатии.
- Молекулярные механизмы генных, хромосомных и геномных мутаций
- Биологический код, его свойства, значение в биосинтезе белка. Взаимодействие кодонов с антикодонами
- Макроэргические соединения. Их роль в клетке
- Характеристика высокоэнергетических фосфатов. Цикл атф-адф
- Тема 5. 6. Разобщение дыхания и синтеза атф
- Глк-глюкоза, Фру-фруктоза, Гал-галактоза
- Синтез глюкозы (глюконеогенез)
- Биосинтез и мобилизация гликогена: последовательность реакций, регуляция гормонами, физиологическое значение.
- Регуляция активности фосфорилазы гормонами.
- Основные пути превращения глюкозы в печени.
- Регуляция обмена углеводов инсулином, глюкагоном, адреналином, кортизолом.
- Пентозофосфатный путь окисления глюкозы, физиологическое значение.
- Гликолипиды и гликопротеины. Представления о строении и функциях их углеводных компонентов.
- I. Структура, классификация и свойства основных липидов организма человека
- Б. Структура и классификация фосфолипидов и сфинголипидов
- Пищевые жиры: норма суточного потребления, переваривание, всасывание продуктов.
- В поддержании гомеостаза холестерола в организме. Биохимия желчнокаменной болезни
- Тема 8. 3. Хиломикроны - транспортная форма экзогенных жиров
- Бета-Окисление жирных кислот. Последовательность реакций. Энергетическое значение.
- Механизмы биосинтеза жирных кислот. Регуляция этого процесса.
- Регуляция синтеза жирных кислот.
- Биосинтез жиров в печени и жировой ткани. Регуляция синтеза жиров
- Депонирование и мобилизация жиров в жировой ткани. Механизм регуляции активности липазы гормонами.
- Фосфолипиды, строение, биологическая роль.
- Холестерин, этапы биосинтеза, биологические функции, регуляция биосинтеза.
- Транспортные формы холестерина. Нарушения транспорта. Семейная гиперхолестеринемия. Атеросклероз.
- Причины и факторы риска развития атеросклероза
- Первичные эндогенные формы истощения
- Представления о биосинтезе фосфолипидов. Липотропные вещества.
- Связь между обменом белков и углеводов
- Влияние инсулина, глюкагона и адреналина на обмен жиров.
- Тема 9. 2. Переваривание белков в желудке и кишечнике, всасывание аминокислот
- Реакции трансаминирования, ферменты, их коферментная группа. Биологическое значение реакций. Определение аминотрансфераз с диагностической целью
- Окислительное дезаминирование аминокислот. Глутаматдегидрогеназа. Значение этой реакции.
- Непрямое дезаминирование аминокислот: последовательность реакций, ферменты, биологическое значение.
Энзимопатии – это группа заболеваний, которые вызваны различными дефектами ферментов. Энзимопатий делятся на: наследственные (первичные) и приобретенные (вторичные).
Наследственные энзимопатии – это заболевания, вызванные наследственными нарушениями биосинтеза ферментов или их структуры и функции.
В норме:

Полное или частичное нарушения биосинтеза ферментов вызывают дефекты генов регуляторных белков, которые контролируют синтез ферментов:

Нарушение структуры и функции ферментов вызывают дефекты генов этих ферментов:

У образовавшегося фермента наблюдаются структурные изменения, которые проявляются в изменении его каталитической активности (как правило, она исчезает), чувствительности к активаторам и ингибиторам, сродству к субстратам, оптимумам рН, температуры. В связи с этим изучением констант фермента является решающим в постановке диагноза врожденных энзимопатий.
Наследственные энзимопатии по типу нарушений метаболизма делят на:
1. нарушения обмена аминокислот: фенилкетонурия, альбинизм, алкаптонурия и др.;
2. нарушения углеводного обмена: галактоземия, наследственная непереносимость фруктозы, гликогенозы;
3. нарушения липидного обмена: липидозы;
4. нарушения обмена нуклеиновых оснований: подагры, синдрома Леш-Нихана и др.;
5. нарушение обмена в соединительной ткани: мукополисахаридозы, хондродистрофия и др.;
6. дефекты ферментов в ЖКТ: муковисцидоз, целиакия, непереносимость лактозы и др.
7. нарушения обмена стероидов и т.д.
В норме метаболический путь протекает следующим образом:

Из-за дефекта в метаболическом пути (цикле, шунте) одного из ферментов в организме происходит накопление промежуточных продуктов (часто токсичных в высоких концентрациях) и дефицит жизненно необходимых конечных продуктов, что приводит к клиническим проявлениям:

Пример: фенилпировиноградная олигофрения – наследственное заболевание, приводящее в раннем детстве к гибели ребенка или к развитию у него тяжелой умственной отсталости.
Причиной заболевания является отсутствие в печени фермента фен-4-монооксигеназы, которая обеспечивает превращение незаменимой аминокислоты Фен в Тир:
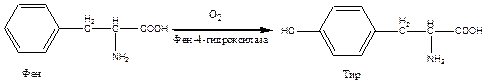
Эта реакция необходима для катаболизма Фен, т.е. удаления его излишков. При отсутствии фен-4-монооксигеназы в организме происходит накопление Фен и превращение его в различные производные: фенилпировиноградную, фенилмолочную и фенилуксусную кислоты.
Фен и его производные в высоких концентрациях токсичны, накапливаясь в тканях, они оказывают на них повреждающее действие. Самой чувствительной к Фен и его производным оказывается нервная ткань детей, она поражается в первую очередь.
Диагноз фенилкетонурия ставят на основании обнаружения Фен в крови или фенилпировиноградной кислоты на пеленках детей. Лечение в основном сводится к исключению из питания ребенка Фен. Для такого ребенка Тир оказывается незаменимой аминокислотой.
Другое тяжелое наследственное заболевание – галактеземия (непереносимость молочного сахара), связано с отсутствием синтеза в печени ферментов, катализирующих превращение галактозы в глюкозу. В результате в раннем возврате происходит накопление в тканях галактозы, приводящее к развитию катаракты, поражению печени, мозга, нередко вызывающее гибель ребенка. Лечение в данном случае сводиться к исключению из диеты молочного сахара.
|