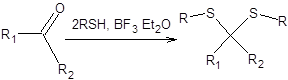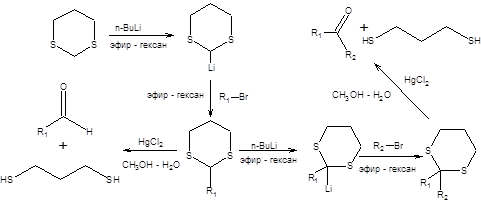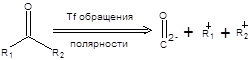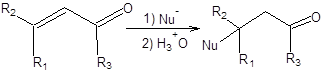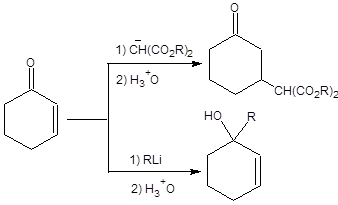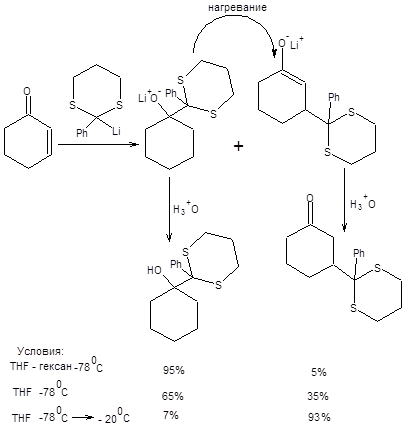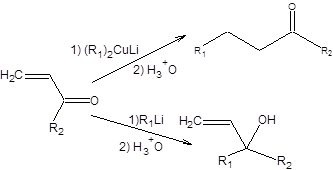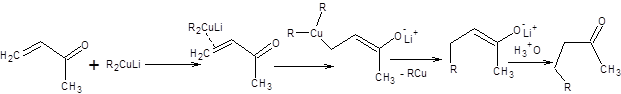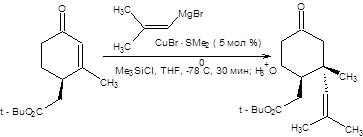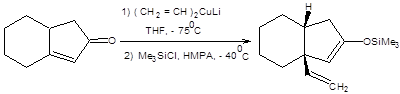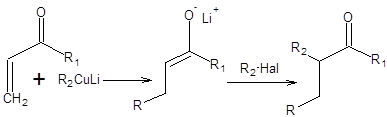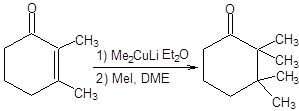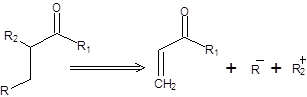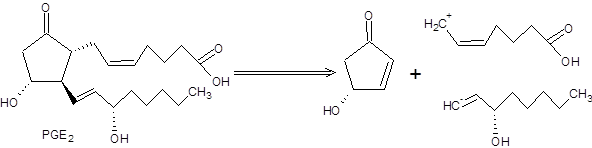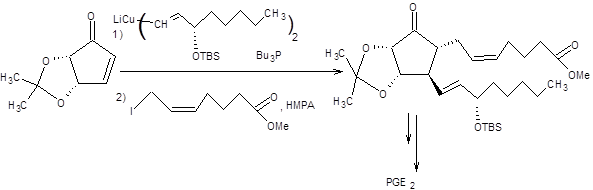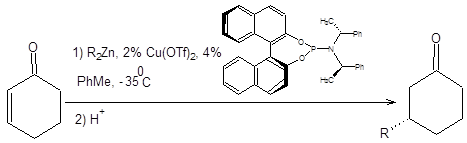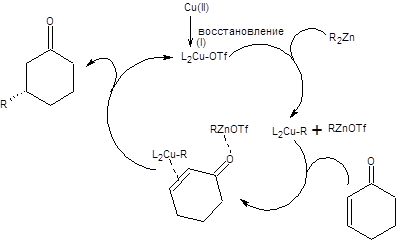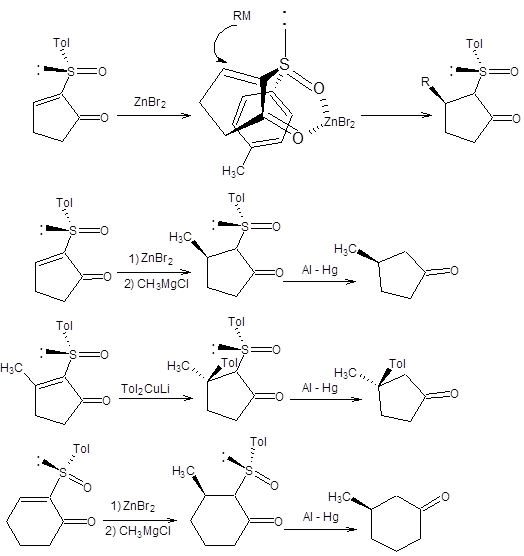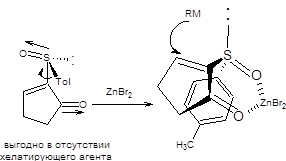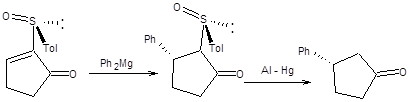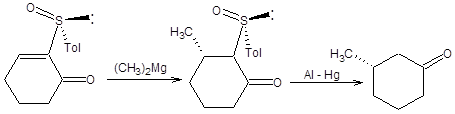Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь FAQ Написать работу КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Метод обращения полярности Кори – ЗеебахаСодержание книги
Поиск на нашем сайте
Альдегиды и кетоны при взаимодействии с тиолами в присутствии кислот Льюиса образуют тиоацетали (тиокетали). Наибольшее распространение в качестве каталитической системы нашел эфират трехфтористого бора. Среди тиоацеталей в современном органическом синтезе совершенно исключительное положение занялшестичленный циклический 1,3-дитиан, получаемый из формальдегида и 1,3-пропандитиола, как показано на схеме 7.32.
Схема 7.32
1,3-Дитиан предоставляет уникальные синтетические возможности, связанные с его способностью нацело превращаться в карбанион под действием сильных оснований, таких как н-бутиллитий. Слабая С–Н-кислотность 1,3-дитиана связана со стабилизацией отрицательного заряда двумя атомами серы. Анион 1,3-дитиана может быть легко алкилирован действием алкилгалогенидов, причем повторное депротонирование, позволяет дважды алкилировать 1,3-дитиан (схема 7.33) [6]. Безусловным достоинством тиоацеталей в синтезе является возможность их трансформации в карбонильные соединения в исключительно мягких условиях катализа ионами ртути.
Схема 7.33
Последовательность стадий превращения альдегидов в производные 1,3-дитиана и их последующего депротонирования приводит к обращению заряда на атоме углерода, т.е. к обращению полярности карбонильной группы. Общая схема метода обращения полярности в терминах ретросинтетического анализа приведена ниже:
Схема 7.34
Метод обращения полярности с успехом применяется для синтеза α-кетоспиртов и β-кетоспиртов при взаимодействии анионов 1,3-дитиана с альдегидами или кетонами и эпоксидами соответственно, а также для синтеза спиртов при восстановлении продуктов конденсации никелем Ренея. Схемы синтеза α-кетоспиртов, β-кетоспиртов и спиртов исходя из 1,3-дитиана приведены ниже:
Схема 7.35 Реакции с участием 1,3-дитиана проводятся в мягких условиях, что позволяет применять их в синтезе полифункциональных субстратов. Список литературы 1. D’Angelo J. // Tetrahedron. 1976. V. 32. P. 2979. 2. Смит В. А., Дильман А. Д. Основы современного органического синтеза. М.: Бином. Лаборатория знаний, 2009. С. 171 - 201. 3. Fleming I., Paterson I. // Sythesis. 1979. P. 736. 4. Schreiber S. L., Wang Z. // J. Am. Chem. Soc. 1985. V. 107. P. 5303. 5. Reetz M. T. et. al. // Chem. Ber. 1980.V. 113. P. 3741. 6. Corey E. J., Seebach D. // Angew. Chem. Int. Ed. Engl. 1965. V. 4. P. 1075.
Лекция 8. Метод сопряженного присоединения и использование енаминов в органическом синтезе
Реакция Михаэля
Реакция Михаэля открывает удобный способ построения углерод – углеродной связи, что может быть использовано, в частности для получения карбонильных соединений. Реакция Михаэля состоит в 1,4-присоединении эквивалентов карбаниов к сопряженным енонам (схема 8.1) [1 – 3].
Схема 8.1
При взаимодействии сопряженных енонов с нуклеофилами, в общем случае, возможно как 1,2-присоединение, так и 1,4-присоединение нуклеофилов. Направление присоединения решающим образом зависит от устойчивости нуклеофильной частицы, природы противоиона и используемого растворителя. Продукт 1,2-присоединения преобладает в условиях кинетического контроля, а продукт 1,4-присоединения в условиях термодинамического контроля. Образованию продукта 1,4-присоединения способствует использование стабилизированных карбанионов в качестве нуклеофилов в сольватирующих растворителях при повышенной температуре и значительной длительности процесса. Продукт 1,2-присоединения соответствует кинетическому контролю и преобладает при пониженной температуре, если реакция проводится с нестабильными карбанионами в малополярных растворителях (схема 8.2).
Схема 8.2
Показана возможность изомеризации продукта 1,2-присоединения в более термодинамически стабильный продукт 1,4-присоединения при повышении температуры, причем увеличение сольватирующей способности растворителя приводит к увеличению доли 1,4-продукта (термодинамического контроля) (схема 8.3) [4].
Схема 8.3
Селективное образование одного из альтернативных продуктов решающим образом зависит от способности первоначально образующегося продукта 1,2-присоединения к обратимой фрагментации с образованием исходных реагентов. Если наблюдается обратимая фрагментация кинетического продукта 1,2-присоединения, то устанавливается равновесие, в котором преобладает термодинамически более устойчивый продукт 1,4-присоединения, что определяет региоселективность реакции. Так как способность продукта 1,2-присоединения к фрагментации возрастает с увеличением температуры, то с ростом температуры его выход уменьшается. В ряде случаев даже при преобладании 1,4-продукта присоединения реакция была фактически необратимой, что говорит о преимущественной нуклеофильной атаке по положению 4 сопряженной еноновой системы. Необратимое присоединение к сопряженным енонам характерно для эквивалентов нестабилизированных карбанионов. В таких случаях особенно существенно влияние природы противоиона на региоселективность присоединения нуклеофилов к сопряженным енонам. Жесткие катионы щелочных металлов (Li+, Na+, Мg2+) обеспечивают атаку по более жесткому электрофильному центру – карбонильной группе, мягкие катионы (Cu+, Cd2+) способствуют атаке по 4-му положению сопряженной еноновой системы. Так, литийалкилы присоединяются по карбонильной группе, а органокупраты по концевому углеродному атому сопряженной еноновой системы. Реактивы Гриньяра обыкновенно испытывают 1,4-присоединение, однако это связано с содержащимися в них примесями ионов d-элементов, в первую очередь железа и меди. Реактивы Гриньяра, полученные из магния сверхвысокой чистоты, присоединяются преимущественно по карбонильной группе. Для осуществления сопряженного присоединения к енонам по Михаэлю с образованием кетонов наибольшее значение имеют органокупраты. Изложенные закономерности можно суммировать следующей схемой, приведенной ниже:
Схема 8.4
Механизм присоединения купратов сложен и до конца не изучен. Принято считать, что изначально происходит координация сопряженного енона с образованием d,π-комплекса купрата, который затем превращается в енолят с последующим быстрым восстановительным элиминированием алкилмеди (схема 8.5).
Схема 8.5
Применение купратных реагентов позволяет достигать высокой степени контроля региоселективности и диастереоселективности реакции. Купратные реагенты, будучи значительных размеров, атакуют сопряженные еноны с наименее затрудненной пространственно стороны, т.е. присоединение органокупратов по Михаэлю – процесс диастереоселективный (схема 8.6) [5, 6].
Схема 8.6
Тандем реакций Михаэля и алкилирования енолятов является одним из наиболее эффективных методов органического синтеза и часто применялся в синтезе природных веществ, в частности простагландинов и может быть представлен общей схемой 8.7.
Схема 8.7
В качестве примеров использования тандема присоединение–алкилирование рассмотрим синтезы, представленные на схеме 8.8.
Схема 8.8 В терминах ретросинтетического анализа тандем реакций присоединения купратов по Михаэлю и последующего алкилирования образующихся енолятов может быть формализован схемой, приведенной ниже:
Схема 8.9
Бесспорным достоинством реализации такого подхода при планировании многостадийных синтезов является автоматическая организация конвергентной схемы синтеза. Тандем реакций Михаэля и алкилирования енолятов стал ключевой стадией синтеза одного из простагландинов PGE2. Ретросинтетический анализ простагландина PGE2 включает расчленение целевой структуры на три блока, каждый из которых синтезировался независимо (схема 8.10) [7]:
Схема 8.10
Сборка целевой молекулы осуществляется путем присоединения соответствующего купрата по Михаэлю с последующим алкилированием полученного енолята, как представлено на схеме 8.11 [8]:
Схема 8.11
Присоединение нуклеофилов по реакции Михаэля позволяет решать сложные задачи построения углеродного скелета, поэтому важность ее осуществления в асимметрическом варианте представляется очевидной. Два последних десятилетия в этом направление проводились интенсивные исследования, итогом которых стало несколько схем, позволяющих проводить каталитическую энантиоселективную реакцию Михаэля. Один из разработанных подходов основан на сопряженном присоединении цинкорганических соединений в условиях катализа трифлатом меди (II). В качестве катализатора асимметрического присоединения по Михаэлю, используется хиральный фосфорсодержащий лиганд, представленный на схеме 8.12. Механизм асимметрического присоединения по Михаэлю включает восстановление меди (II) до меди (I) с последующим образованием медьорганического соединения, которое и участвует в ключевом акте сопряженного присоединения. Механизм энантиоселективного присоединения органокупратов по Михаэлю представлен на схеме 8.12 [9].
Схема 8.12
Другой подход к проведению асимметрического присоединения по Михаэлю основан на использовании энантиомерно чистых сульфоксидов и надежно зарекомендовал себя в синтезе 3-замещенных циклоалканонов [10]. Исходные энантиомерно чистые 2-толилсульфинил-2-циклоалкеноны получают из сопряженных циклоалкенонов. В качестве источника хиральности в синтезах 2-толилсульфинил-2-циклоалкенонов используется (-)-ментол. Синтез хирального 2-толилсульфинил-2-циклопентанона приведен на схеме 8.13. Аналогичным образом могут быть получены другие хиральные 2-толилсульфинил-2-циклоалкеноны.
Схема 8.13
Сопряженное присоединение к 2-толилсульфинил-2-циклоалкенонам наблюдается не только для диалкилкупратов, но и для реактивов Гриньяра ввиду высокой стабильности образующегося карбаниона, стабилизированного одновременно двумя электроноакцепторными заместителями. Схема 8.14 В зависимости от условий проведения сопряженного присоединения из одного энантиомера 2-толилсульфинил-2-циклоалкенона может быть получен с высокой оптической чистотой один из двух возможных энантиомерных 3-замещенных циклоалканонов. Если реакция проводится в присутствии хелатирующего агента, например, бромида цинка, то исходный 2-толилсульфинил-2-циклоалкенон фиксируется в конформации, показной на схеме 8.14. Нуклеофильная атака реактивом Гриньяра осуществляется с наименее затрудненной пространственно стороны. Модель хелатирования справедлива также в случае присоединения диалкилкупратов (схема 8.14). Если сопряженное присоединение проводится под действием диалкилмагния без предварительного хелатирования, то стереохимический результат присоединения будет обратным, показанному на схеме8.77. Причиной различия стереохимических результатов присоединения к хелатированной и нехелатированной формам 2-толилсульфинил-2-циклоалкенонов являются различия устойчивых относительных ориентаций сульфоксидной группы и кетогруппы в переходном состоянии. В отсутствии хелатирующего агента кетогруппа и сульфоксидная группа, ориентированы по отношению к друг другу противоположными концами диполей, что наиболее выгодно энергетически. В присутствии хелатирующего агента геометрия исходного хирального сульфоксида изменяется, как показано на схеме 8.15. Примеры реализации асимметрического сопряженного присоединения диалкилмагниевых соединений к энантиомерночистым 2-толилсульфинил-2-циклоалкенонам без предварительного хелатирования приведены на схеме 8.15.
Схема 8.15 Использование хиральных сульфоксидов открывает перспективный путь к осуществлению асимметрического присоединения по Михаэлю [11] и находит применение в ряде синтезов природных соединений.
|
||
|
|
Последнее изменение этой страницы: 2021-05-27; просмотров: 747; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 216.73.216.153 (0.007 с.) |