Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь FAQ Написать работу КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Тромбоцитарные факторы свертывания (арабская нумерация)Содержание книги
Поиск на нашем сайте ТРОМБОЦИТАРНЫЕ ФАКТОРЫ СВЕРТЫВАНИЯ (арабская нумерация) Фактор 1 – тромбоцитарный акцелератор-глобулин, идентичен фактору V Фактор 2 – акцелератор тромбина, фибринопластический фактор (ускоряет превращение фибриногена) Фактор 3 – тромбоцитарный тромбопластин, частичный тромбопластин Фактор 4 – антигепариновый фактор Фактор 5 – свертываемый фактор (иммунологически идентичен фибриногену) Фактор 6 – тромбостенин Фактор 7 – тромбоцитарный котромбопластин Фактор 8 – антифибринолизин Фактор 9 – фибринстабилизирующий фактор, по действию соответствует фактору XIII Фактор 10 – 5-гидрокситриптамин, серотонин Фактор 11 – аденозиндифосфат (АДФ) МЕХАНИЗМЫ ГЕМОСТАЗА Первичный механизм гемостаза – микроциркуляторный, сосудисто-тромбоцитарный. Он осуществляется в капиллярах, венозных и артериальных сосудах до 100-200 мкм в диаметре. Непосредственно участвуют в этом процессе тромбоциты и сосудистый эндотелий, реакции между которыми происходят в сосудах микроциркуляторного русла. Нарушения этого механизма клинически определяют 80% кровотечений и 95% тромбообразования.
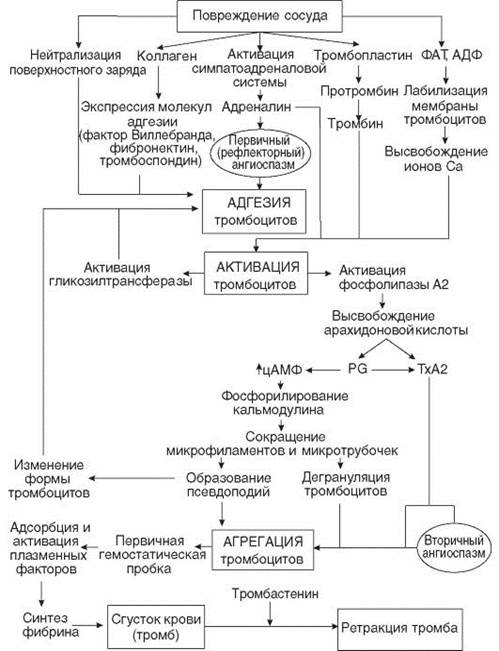
Вторичный механизм гемостаза – макроциркуляторный, гемокоагуляционный. Он реализуется с участием системы свертывания крови. Начинается на основе первичного гемостаза, следует за ним. Благодаря вторичному гемостазу образуется красный кровяной тромб, состоящий из фибрина и форменных элементов крови. Он обеспечивает окончательную остановку кровотечения из поврежденных макрососудов (более 200 мкм в диаметре). СХЕМА СОСУДИСТО-ТРОМБОЦИТАРНОГО ГЕМОСТАЗА (Е.Д. Гольдбергу, 2009 )
СХЕМА КОАГУЛЯЦИОННОГО ГЕМОСТАЗА (по Е. П. Иванову, 1979, 1980)
ФИЗИОЛОГИЧЕСКИЕ АНТИКОАГУЛЯНТЫ Первичные (предшествующие, имеются в крови до начала свертывания крови)
Вторичные антикоагулянты (образуются в процессе свертывания крови или фибринолиза)
НАРУШЕНИЕ СИСТЕМЫ ГЕМОСТАЗА В зависимости от того, в каком звене системы гемостаза произошли нарушения, все заболевания этой группы подразделяют на следующие типы:
Проявления тромбоцитопений Костный мозг: – гиперплазия с увеличением числа мегакариобластов и мегакариоцитов (при повышенном разрушении или генерализованном «потреблении» тромбоцитов) – гипоплазия (у пациентов с лейкозами, лучевой болезнью, метастазами опухолей в костный мозг) – снижение содержания гликогена и активности ЛДГ, ГЛ-6-ФДГ в мегакариобластах и мегакариоцитах, что уменьшает продолжительность жизни тромбоцитов Периферическая кровь: – уменьшение числа тромбоцитов и увеличение их размеров при обычно нормальном количестве эритроцитов, Hb, лейкоцитов – при выраженном геморрагическом синдроме возможно развитие анемии.
ИММУННЫЕ ТРОМБОЦИТОПЕНИИ Иммунная тромбоцитопеническая пурпура (ИТП) — группа заболеваний, объединенных единым патогенезом. Выделяют следующие формы иммунной тромбоцитопенической пурпуры: аутоиммунные — аутоАТ образуются к неизмененным Tr (болезнь Верльгофа). гетероиммунные — АТ образуются против измененных антигенов Tr. трансиммунные — при трансплацентарной передаче плоду антитромбоцитарных АТ от матери с болезнью Верльгофа. изоиммунные — при несовместимости по антигенам тромбоцитов матери и плода или донора и реципиента при гемотрансфузиях. гаптеновые — связанны с фиксацией на мембране Tr гаптенов (ЛС, вирусы). Патогенез: снижение продолжительность жизни тромбоцитов до нескольких часов, вследствие образования к ним антител (АТ) и разрушение комплементом или фагоцитами. Проявления: – множественные петехии – синяки, провоцируемые незначительными ушибами – спонтанно возникающие кровотечения (носовые, маточные, реже из мочевыводящих путей и ЖКТ); при повторных кровотечениях — ЖДА – возможны кровоизлияния в мозг
Тромбастения Гланцмана Этиология: отсутствие или дефект мембранного рецептора к фибриногену и гликопротеинам IIb–III a Патогенез: снижение интенсивности связывания фибриногена с мембраной Tr, нарушение агрегации Tr Проявления: – петехиально-экхимозный тип кровоточивости – склонность к кровотечению из слизистых (носовые, маточные, кровоизлияния в склеру и сетчатку глаза) – длительные кровотечения – после удаления зуба, ЛОР-операций Болезнь Бернара-Сулье (макроцитарная тромбодисрофия, синдром гигантских тромбоцитов) Этиология: в мембране Tr отсутствует специфический гликопротеины, взаимодействующий с комплексаФВ-ФVIII, ФV, ФIX и ристоцетином, также повышено содержание сиаловых кислот, снижен электрический заряд. Патогенез: нарушение адгезионных свойств Tr, снижение продолжительности жизни Tr при нормальной продукци в КМ → умеренная тромбоцитопения. Основной морфологический критерий: наличие в крови гигантских Tr, достигающих 6–8 мкм Проявления Кровоточивость петехиального типа (тяжесть кровоточивости зависит от содержания аномальных тромбоцитов:чем выше их %, тем тяжелее и опаснее протекает геморрагический синдром). Болезнь Виллебранда Фактор Виллебранда (ФВ) продуцируется эндотелиальными клетками и мегакариоцитами, где он хранится в органеллах. В эндотелиальных клетках фактор Виллебранда хранится в тельцах Вейбла-Палада, а в тромбоцитах — в α-гранулах. Болезнь Виллебранда бывает наследственной и приобретенной. Этиология: дефицит и/или дефект структуры фактора Виллебранда (ФВ) Патогенез: нарушение адгезии Tr к коллагену сосудистой стенки и снижение интенсивности образования комплекса ФВ-ФVIII. При отсутствии фактора Виллебранда фактор VIII подвергается ускоренному разрушению в крови, что и обусловливает его дефицит и связанную с ним спонтанную гематомную кровоточивость. Таким образом, болезнь Виллебрандахарактеризуется нарушением тромбоцитарного и коагуляционного гемостаза. Проявления: – экхимозные, реже – гематомные кровоизлияния – кровоточивость слизистых оболочек – высокий риск профузных кровотечений при хирургических вмешательствах
КОАГУЛОПАТИИ ( патологии свертывающей системы крови) Коагулопатии бывают наследственные (гемофилии А, В, С, парагемофилии, «женская гемофилия») и приобретенные (ДВС-синдром). НАСЛЕДСТВЕННЫЕ КОАГУЛОПАТИИ Гемофилия А Этиология: дефицит фактора VIII (антигемофильный глобулин). Наследуется рецессивно, сцепленно с Х -хромосомой. Болеют лица мужского пола (10 на 100 тыс. мужчин) Патогенез: дефицит приводит к резкому увеличению времени образования протромбиназного комплекса, что сопровождается длительным, практически не прекращающимся кровотечением при незначительной травматизации сосудов (прикусывание языка, ушибы). Проявления Для гемофилии А характерен гематомный тип кровоточивости. При легкой форме кровотечения возможны лишь при значительных травмах или оперативных вмешательствах, протекает субклинически и часто не диагностируются. При тяжелой или очень тяжелой форме рецидивирующие кровоизлияния в крупные суставы (гемартрозы), приводят к анкилозированию, крупные меж- и внутримышечные, забрюшинные гематомы с последующей деструкцией мягких тканей, тяжелые и частые спонтанные кровотечения, упорные рецидивирующие желудочно-кишечные и почечные кровотечения. Гемофилия В (болезнь Кристмаса) Этиология: дефицит фактора IX (фактор Кристмаса). Наследуется рецессивно, сцепленно с Х -хромосомой. Патогенез: дефект приводит к значительному замедлению формирования протробиназного комплекса → развитие кровоточивости гематомного типа. Проявления Клиническая картина характеризуется кровотечениями (гемартрозы, гематомы), но частота их в 5 раз ниже, чем при дефиците фактора VIII. Гемофилия С Этиология: дефицит фактора ХI (плазменный предшественник тромбопластина, фактор Розенталя). Наследуется аутосомно-рецессивно. Патогенез: изолированное нарушение внутреннего механизма процесса свертывания крови. Проявления У гетерозигот кровотечения бывают незначительны. У гомозигот с дефицитом ФХI осложнений, связанных с кровоточивостью —немного, но при травме или хирургическом вмешательстве не исключено возникновение сильного кровотечения с формированием гемартроза и гематомы. Парагемофилия (болезнь Оурена) Этиология: дефицит фактора V(проакцелерин, церулоплазминоподобный связывающий белок). Наследуется аутосомно-рецессивно и аутосомно-доминантно. Проявления Для заболевания характерен геморрагический синдром: отмечаются петехии, эсхимозы, кровоподтеки, носовые, десневые, желудочно-кишечные кровотечения, меноррагии. У пациентов при выраженных формах заболеваний часты длительные кровотечения после удаления зубов, при травмах, порезах.
«Женская гемофилия» встречается крайне редко (описано около 50 случаев). Это разнородные в генетическом отношении заболевания.
Основные варианты «женской гемофилии»: 1. Больные с нормальным женским набором половых хромосом (ХХ) и двойным наследованием истинной гемофилии — возникает у девочек, отцы которых болеют гемофилией, а матери являются кондукторами заболевания (причина — браки между кровными родственниками). 2. Больные с нормальным набором половых хромосом (ХХ) и односторонней гемофилической наследственностью. 3. Больные с неполным набором хромосом и одной Х-хромосомой (кариотип ХО). Могут болеть такой же тяжелой формой гемофилии, как мужчины из той же семьи. 4. Гемофилия у лиц «женского пола» с тестикулярной феминизацией (имеют в соматических клетках мужской набор хромосом — ХY). 5. Аутосомно-доминантные формы дефицита VIII фактора (в этой группе необходимо исключать болезнь Виллебранда). ПИОБРЕТЕННЫЕ КОАГУЛОПАТИИ Принципы классификации 1. по течению: – острый; – подострый; – хронический; – рецидивирующий; – латентный.
2. по локализации процесса: – диссеминированное внутрисосудистое свертывание крови (на уровне целого организма); – локализованное внутрисосудистое свертывание крови (в пределах органа или его части). 3. по тяжести процесса: – компенсированный; – субкомпенсированный; – декомпенсированный Этиология – все виды шока, травмы (например, при краш-синдроме) – массивные деструкции и некрозы в органах – травматичные хирургические вмешательства – термические и химические ожоги – острый внутрисосудистый гемолиз (при переливании несовместимой крови и массивных гемотрансфузиях) и хроническом гемолизе – тяжелые инфекции и сепсис – некоторые формы акушерско-гинекологической патологии (эмболия сосудов матери околоплодными водами, криминальные аборты, инфекционно-септические осложнения в родах и при прерывании беременности и др.) – терминальные состояния – укусы ядовитых змей. – персистирующие бактериальные и вирусные инфекции – СКВ – злокачественные опухоли – обезвоживание организма – массивный контакт крови с чужеродной поверхностью (экстракорпоральное кровообращение, хронический гемодиализ) Основные звенья патогенеза ДВС-синдрома: – начальная активация свертывающей системы и тромбоцитов эндогенными факторамит (тканевым тромбопластином, лейкоцитарными протеазами, продуктами распада тканей, опухолевыми прокоагулянтами) – персистирующая тромбинемия с повышением уровня ее маркеров в крови; – истощение системы физиологических антикоагулянтов (снижением содержания в плазме антитромбина III, протеина С, плазминогена и повышение уровня тромбомодулина) – системное поражение сосудистого эндотелия и снижение его антитромботического потенциала; – образование микросгустков крови и блокада микроциркуляции в органах-мишенях с развитием в них дистрофических и деструктивных нарушений; – активация фибринолиза в зоне блокады микроциркуляции и истощение его резервов в общей циркуляции; – потребление факторов гемокоагуляции и тромбоцитопения (и тромбоцитопатия) потребления, приводящие к системной кровоточивости и терминальной гипокоагуляции вплоть до полной несвертываемости крови (геморрагическая фаза синдрома); – нарушение барьерной функции слизистой оболочки желудка и кишечника с трансформацией асептического ДВС-синдрома в септический; –вторичная тяжелая эндогенная интоксикация. Стадии 1 стадия: гиперкоагуляция и агрегация тромбоцитов 2 стадия: переходная (коагулопатия потребления без активации фибринолиза) 3 стадия: гипокоагуляция (коагулопатия потребления с активацией фибринолиза) 4 стадия: восстановительная (остаточные тромбозы и блокады сосудов) Проявления Для ДВС-синдрома характерны симптомы полиорганной недостаточности в результате поражения и дисфункции органов-мишеней. 1. Острая легочная недостаточность (вплоть до легочного дистресс-синдрома). 2. Острая почечная или гепаторенальная недостаточность (снижение диуреза и уремия). 3. Церебральная симптоматика, связанная с ишемией мозга. 4. Поражение слизистой оболочки желудка и кишечника (обильные кровотечения, острые гипоксические язвы, нарушение барьерной функции слизистой оболочки и трансформация асептических форм ДВС в септико-токсические формы). 5. Надпочечниковая, или плюригландулярная, эндокринная недостаточность с нестабильной гемодинамикой. 6. Синдром системной воспалительной реакции с накоплением в крови цитокинов и других метаболитов.
ТРОМБОЦИТАРНЫЕ ФАКТОРЫ СВЕРТЫВАНИЯ (арабская нумерация) Фактор 1 – тромбоцитарный акцелератор-глобулин, идентичен фактору V Фактор 2 – акцелератор тромбина, фибринопластический фактор (ускоряет превращение фибриногена) Фактор 3 – тромбоцитарный тромбопластин, частичный тромбопластин Фактор 4 – антигепариновый фактор Фактор 5 – свертываемый фактор (иммунологически идентичен фибриногену) Фактор 6 – тромбостенин Фактор 7 – тромбоцитарный котромбопластин Фактор 8 – антифибринолизин Фактор 9 – фибринстабилизирующий фактор, по действию соответствует фактору XIII Фактор 10 – 5-гидрокситриптамин, серотонин Фактор 11 – аденозиндифосфат (АДФ)
|
||||||||||||||||||||||||||||||||||||||||
|
|
Последнее изменение этой страницы: 2017-01-27; просмотров: 323; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 216.73.216.153 (0.007 с.) |