Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь FAQ Написать работу КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Органические реагенты в химическом анализеСодержание книги
Поиск на нашем сайте
План: 1. Определение органических реагентов (ОР). 2. Преимущества ОР перед неорганическими реагентами 3. Классификация ОР 4. теория функционально-аналитических (ФАГ) и аналитико-активных групп (ААГ). 5. теория аналогий 6. Применение ОР в анализе.
1. Органические реагенты – это органические соединения, которые используются для обнаружения, разделения и определения как органических, так и неорганических ионов или молекул. Органические реагенты и органические соединения – разные понятия. Органические реагенты – более широкое понятие. Первый органический реагент предложен в 1879 г. Гриссом (смесь сульфониловой кислоты с α-нафтиламином). В 1886 г Ильинский использовал α-нитрозо-β-нафтол в качестве реагента на кобальт. Чугаев применил диметилглиоксим для обнаружения и определения никеля (1905 г.). Чугаев не только предложил новые органические реагенты, но и разработал теоретические основы их действия. 2. Преимущества ОР перед неорганическими: а) высокая чувствительность и избирательность; б) интенсивная окраска продуктов реакций с ОР; в) малая растворимость в воде; г) многообразие ОР. 3. Классификацию ОР проводил Кульберг Л.М. по механизму действия и характеру конечного продукта. I. Органические реагенты, приводящие к образованию простых солей (Ca2+ с H2C2O4). II. Органические реагенты, приводящие к образованию ВКС; III. ОР, действие которых основано на реакциях окисления-восстановления: Хромофором является цепочка сопряженных двойных связей (хиноидное положение) IV. ОР, приводящие к синтезу новых соединений. Для обнаружения NO2- используется реактив Грисса. Синтез идет в две стадии: 1) реакция диазотирования В качестве диазотирующего вещества используется HNO2. Проводят диазотирование сульфониловой кислоты.
2) реакция азосочетания
Хромоформом является азо-группа. V. ОР, действие которых основано на адсорбционном процессе. В качестве примера рассмотрим действие адсорбционного индикатора флуоресцеина в аргентометрии. Сущность аргентометрии заключается в том, что Cl- -ионы титруют раствором АgNO3. титрование – это процесс прибавления раствора с точно известной концентрацией к определяемому веществу. Для фиксирования точки эквивалентности (ТЭ) (момента титрования, когда количество определяемого вещества эквивалентно количеству титранта) используют флуоресцеин, который представляет собой слабую органическую кислоту: HФ ↔H+ + Ф-. К раствору, содержащему Cl-, прибавляют АgNO3, образуется AgCl. До ТЭ на поверхность осадка адсорбируются Cl- -ионы, осадок заряжается отрицательно. После ТЭ в растворе в избытке будут ионы Ag-, поверхность осадка заряжается положительно. Осадок адсорбирует анионы осадка и приобретает розовую окраску. VI. ОР, приводящие к образованию разнолигандных комплексов, например, Fe3+ с сульфосалициловой кислотой в зависимости от pH образует комплексы различного состава, имеющие разную окраску. При pH ~4-8 комплекс фиолетового цвета, при pH~ 8-11 комплекс желтого цвета. 4. Кульберг делит аналитический центр на 2 группы: ФАГ и ААГ. ААГ – это группа атомов, которая существенно не влияя на механизм реакции изменяет характер конечного продукта (влияет на чувствительность, интенсивность окраски и растворимость осадка). К ААГ относятся: -SO3H; -COOH; -NH2; –OH; -NO2 и др.
Примеры: Диметилглиоксим
Арсеназо I
За счет удвоения Савин получил реагент – арсеназо III, который взаимодействует с ионами более 30 металлов с образованием окрашенных комплексов.
Ион Zn4+ в 10М HCl с арсеназо образует ВКС зеленого цвета. Комплексы с остальными ионами металлов в этих условиях разрушаются. Арсеназо III является специфичным реагентом на Zn4+ в 10М HCl. 5. Основателем теории аналогий является профессор Кузнецов. Он сравнивает поведение ОР со свойствами его простейших неорганических аналогов. ОР типа ROH сравнивает с H2O RNH2 c NH3 RSH c H2S Реагенты типа ROH должны взаимодействовать со всеми ионами, которые подвергаются гидролизу. Причем взаимодействие происходит при тех же значениях pH, при которых происходит гидролиз. pH образования гидроксидов Сильно-кислая среда Zn4+, Th4+, Кислая Al3+, Fe3+ Слабо-кислая, нейтральная Co2+, Zn2+, Fe2+ Щелочная Ca2+, Mg2+ Реагенты типа RSH взаимодействуют с теми ионами, которые образуют малорастворимые сульфиды (Cu2+, Hg2+, Pb2+ и др.). Теория Кузнецова не учитывает то обстоятельство, что ОР имеют более сложное строение, чем неорганические реагенты. 6. ОР применяются: а) для обнаружения ионов; б) для разделения ионов (дитизон, 8-оксихинолин, пиридин-азо-нафтол); в) для определения ионов (гравиметрическим методом Ni2+ определяют с диметилглиоксимом, комплексонометрическим методом Ni2+ определяют мурексидом). ЛЕКЦИЯ №5 КИСЛОТНО-ОСНОВНЫЕ РЕАКЦИИ 1. недостатки теории Аррениуса 2. Протолитическая теория кислот и оснований Бренстеда-Лоури. а) кислоты, основания, амфолиты; б)сопряженные кислотно-основные пары и их взаимодействие; в) перенос протонов, взаимодействие двух кислотно-основных пар; г) классификация растворителей по донорно-акцепторным свойствам; д) нивелирующий и дифференцирурующий эффекты растворителя; е) константы кислотности и основности, связь между ними.
1. Первой теорией кислот и оснований была теория Аррениуса, в основе которой лежит электролитическая диссоциация веществ в водных растворах. Недостатками этой теории являются: 1) не учитывается влияние растворителя в реакциях кислот и оснований; 2) теория применима только к водным растворам; 3) протон не может существовать в свободном виде, как это требует теория.
2. Всеобщее признание получила протолитическая теория кислот и оснований Бренстеда-Лоури. Кислоты – соединения, способные отдавать протон. Основания – соединения, способные присоединять протон. Кислотами и основаниями могут быть не только молекулярные частицы, но и заряженные. Молекулярные кислоты: HCl = H+ + Cl- CH3COOH ↔CH3COO- + H+ H2O ↔ H+ + OH- Заряженные кислоты: NH4+ ↔ NH3 + H+ H2PO4- ↔ H+ + HPO42- Молекулярные основания: NaOH ↔ Na+ + OH- OH- + H+ ↔H2O Заряженные основания: CH3COO- + H+ ↔ CH3COOH HPO42- + H+ ↔ H2PO4- [Al(H2O)5OH]2+ + H+ ↔ [Al(H2O)6]3+
Амфолиты – вещества, способные как присоединять, так и отдавать протон.
H2O ↔ H+ + OH- H2O + H+ ↔ H3O+ NH3 ↔ H+ + NH2- NH3 + H+ ↔ NH4+ Кислоты, основания, амфолиты называются протолитами. По теории Аррениуса кислоты и основания не связаны между собой, по теории Бренстеда-Лоури, они связаны между собой. Кислота ↔ протон ↔ основание В реальных условиях эти полуреакции не могут протекать: HA ↔ H+ + A- B+ H+ ↔ BH+ Протон в растворе в свободном виде не может существовать, протон отщепляется в том случае, если в системе есть основание, которое присоединяет этот протон. Происходит перенос протона от одной кислотно-основной пары к другой. HA + B ↔ A- + BH+ кис.I осн. II осн. I кис. II Протолитическая реакция представляет собой взаимодействие двух кислотно-основных пар, одну кислотно-основную пару представляет протолит, другую – растворитель. Направление переноса протона зависит от донорно-акцепторных свойств вещества и растворителя. CH3COOH + H2O ↔ CH3COO- + H3O+ Акцепторные свойства у H2O выражены сильнее, чем у CH3COOH, среда кислая NH3 + H2O ↔ NH4+ +OH- Акцепторные свойства у NH3 выражены сильнее, чем у H2O. Протон переносится от H2O к NH3, среда щелочная. По теории Бренстеда-Лоури диссоциация кислот и оснований предсталяет кислотно-основное взаимодействие, которое осуществляется за счет переноса протона от кислоты к основанию и образуются новые кислоты и основания. По кислотно-основным свойствам растворители делят на активные, неактивные (апротонные).
Апротонные растворители не обладают ни кислотными, ни основными свойствами. К ним относятся CCl4, CHCl3, C6H6, C6H14, CS2, C6H12. Активные растворители делятся на протогенные, протофильные, амфипротные.
Протогенные (кислотные) растворители способны к отдаче протона (H2SO4, CH3COOH, ClCH2COOH, H3PO4 и т.д.
Протофильные (основные) растворители способны к присоединению протона (NH3, простые эфиры, (C2H5)2NH, C5H5N).
Амфипротные растворители способны как к отдаче, так и присоединению протона (H2O, спирты, карбоновые кислоты, кетоны) Для амфипротных растворителей характерна реакция автопротолиза.
Автопротолиз – самоионизация растворителя, когда одна молекула растворителя проявляет свойства кислоты, другая – основания H2O + H2O ↔ H3O+ + OH- В общем виде 2SH ↔SH2+ + S- лионий хелат
KSH определяет протяженность шкалы pH. pH – это отрицательный логарифм активности иона лиония:
pH – это отрицательный логарифм активности иона гидроксония:
Для разбавленных растворов активность можно заменить концентрацией, тогда pH = -lg[H+]. Сила кислоты и основания в значительной степени зависит от кислотно-основных свойств растворителя. Силу протолитов и их свойства сравнивают по отношению к одному и тому же растворителю (например, к H2O). В воде такие кислоты, как HClO4, HCl, HNO3, H2SO4 невелируются и имеют одинаковую силу на уровне иона H3O+ HClO4 + H2O ↔ H3O+ + ClO4- HCl + H2O ↔ H3O+ + Cl- HNO3 + H2O ↔ H3O+ + NO3- H2SO4 + H2O ↔ H3O+ + HSO4- У HCOOH акцепторные свойства почти такие же, как у воды. Поэтому протон не переносится. HCOOH в водном растворе является слабой кислотой, может существовать в молекулярной форме и в виде ионов. Вместо H2O возьмем другой растворитель, например, NH3. У него акцепторные свойства сильнее выражены, чем у H2O. NH3 может забирать протон и у HCOOH. В жидком NH3 сила кислот нивелируется до силы NH4+, более слабого, чем H3O-. HClO4 + NH3 ↔ NH4+ + ClO4- HCl + NH3 ↔ NH4+ + Cl- HCOOH + NH3 ↔ NH4+ + HCOO- Чем сильнее выражены акцепторные свойства ратсворителя, тем на более широкий круг распространяется его нивелирующее действие. В ледяной уксусной кислоте HClO4 легче отдает протон, чем HCl. (KHClO4 = 10-4, KHCl =10-7). Выводы: 1. Кислоты протолиты нивелируются в основных растворителях, дифференцируются – в кислотных растворителях. 2. Основания – протолиты нивелируются в кислотных растворителях, дифференцируются - в основных. Силу кислот и оснований характеризуют константами кислотности и основности. HA + H2O ↔ H3O+ + A-
A-+ H2O ↔ HA + OH-
Из (2) находим aHA и подставляем в (1):
ЛЕКЦИЯ №6 БУФЕРНЫЕ РАСТВОРЫ
План: 1. Определение буферных растворов. Типы буферных растворов. 2. Механизм буферного действия. 3. Вычисление pH буферных растворов 4. Буферная емкость 5. Применение в анализе
1. Буферными называются растворы, которые не изменяют заметно pH при разбавлении и при добавлении небольших количеств растворов сильных кислот и оснований. Буферный раствор представляет собой сопряженную кислотно0основную пару. Типы буферных растворов: а) ацетатный –CH3COOH и CH3COONa б) аммонийный – NH3∙H2O и NH4Cl в) формиатный – HCOOH и HCOONa г) фосфатный – Na2HPO4 и NaH2PO4 д) карбонатный – Na2CO3 и NaHCO3 е) индивидуальные соединения – раствор буры содержит эквимолярные количества H3BO3 и сопряженного основания BO2-; - концентрированные растворы сильных кислот и оснований. Сопряженными системами являются H3O+/H2O – для сильных кислот; OH-/H2O – для сильных оснований.
2. Механизм буферного действия состоит в том, что вводимая сильная кислота или сильное основание связывают компоненты буферной смеси в малодиссоциирующие соединения. CH3COOH + NaOH ↔ CH3COONa + H2O CH3COOH + OH- ↔ CH3COO- + H2O CH3COONa + HCl ↔ CH3COOH + NaCl CH3COO- + H+ ↔ CH3COOH Суммарная концентрация CH3COONa и CH3COOH сохраняется, а меняется только соотношение При разбавлении одновременно действуют два взаимно компенсирующих друг друга эффекта. За счет разбавления уменьшается концентрация кислоты и увеличивается степень диссоциации CH3COOH.
3. Вывод формулы
Эти две формулы практически ничем не отличаются. Для водных растворов удобнее пользоваться уравнением Аррениуса. 4. Чтобы буферного раствора не изменился, можно к нему добавлять только определенное количество сильной кислоты и сильного основания. Буферный раствор обладает буферной емкостью. Буферная емкость – число молей эквивалентов сильной кислоты или сильного основания, которые нужно добавить, чтобы pH раствора изменился на единицу. Буферная емкость определяется по формуле:
π зависит от суммарной концентрации компонентов и соотношения их концентраций. Чем больше концентрация компонентов, тем больше буферная емкость.
Тема: Окислительно-восстановительные реакции в аналитической химии. План: 1. Стандартный и формальный потенциалы. 2. Уравнение Нернста. 3. Влияние различных факторов на величину потенциала и направление реакции. 4. Константа равновесия и ее связь со стандартным потенциалом (самостоятельно). Для оценки окислительно-восстановительной способности окислителей и восстановителей служит потенциал. Потенциал является мерой сродства к электрону. Чем больше сродство к электрону, тем сильнее выражены окислительные свойства. В процессе окислительно-восстановительных реакций (ОВР) накапливаются как окислители, так и восстановители, т.е. образуется сопряженная окислительно-восстановительная пара. Например, Br2 + 2ē ↔ Br2/ 2Br-. Происходит перенос электрона от восстановителя к окислителю. Определить абсолютное значение потенциала отдельной окислительно-восстановительной пары невозможно, поэтому потенциализмеряют относительного стандартного водородного электрода (СВЭ). СВЭ представляет собой платиновую пластинку, покрытую мелкораздробленной платиновой чернью, и опущенную в 1 н раствор H2SO4. Через чернь пропускают газообразный H2 под давлением 1 атм. В основе работы СВЭ лежит реакция: 2H+ +2ē ↔ H2. Потенциал у СВЭ условно принят равным нулю. Чтобы получить стандартные условия выбираем систему, у которой aок = aвос = 1 моль/л и измеряем. Потенциал системы, в которой aок = aвос = 1 моль/л, и измеренный относительно СВЭ называется стандартным (Е°). Е° принимать знаки «+» и «-». Знаки при потенциалах показывают, окислителем или восстановителем является данная пара по отношению к СВЭ. Если знак (+), то система является окислителем по отношению к СВЭ. Если знак (-), то система является восстановителем по отношению к СВЭ. Чем выше положительное значение потенциала системы, тем большей окислительной способностью обладает система. Чем более отрицательное значение потенциала системы, тем большей восстановительной способностью обладает система. Система с большим значением потенциала является окислителем по отношению к системе с меньшим значением потенциала. Чем больше разность потенциалов двух пар, тем менее обратима реакция. Чтобы реакция протекала количественно (на 99,9%), разность потенциалов должна быть ∆E ≥ 0.3 B. В реальных условиях aок и aвос форм отличаются от единицы. Потенциал системы, в которой активности aок и aвос форм отличается от единицы называется равновесным (EOx/Red). Зависимость равновесного потенциала от aок и aвос форм выражается через уравнение Нернста:
EOx/Red зависит от природы растворителя, температуры, γ, α. a = γ∙ c∙α Тогда Формальный потенциал – это потенциал системы, в которой COx = CRed = 1моль/л. E°/ зависит от природы, температуры, I и α. Какую формулу используют в расчетах?
Обычно мы работаем с разбавленными растворами, и при отсутствии глубокопротекающих конкурирующих реакций, используем формулу:
На величину потенциала влияет концентрация ионов водорода. При этом различают три случая: 1) H+ непосредственно входит в уравнение Нернста; MnO4- +8H+ +5ē = Mn2+ + 4H2O
2) H+ непосредственно не участвует в реакциях окисления-восстановления, но вызывает конкурирующую реакцию протонирования, тем самым влияет на величину потенциала. S кислой среде протонируется S2- + H+→ HS- → H2S Уравнение материального баланса:
H+ не влияет на величину потенциала, если в ОВР не участвует кислородсодержащий окислитель и отсутствуют конкурирующие реакции. На величину потенциала влияет конкурирующая реакция осадкообразования. Например, рассмотрим иодометрическое определение меди.
Система йода является окислителем по отношению к системе меди. По стандартным потенциалам должна идти реакция: Cu2+ + I2 → Cu2+ + 2I- На самом деле реакция идет в обратном направлении, что связано с образованием малорастворимого соединения CuI.
Потенциал системы за счет образования осадка CuI увеличивается.
Протекает реакция: 2Cu2+ +4I- = 2CuI↓ + I2 На величину потенциала влияет конкурирующая реакция комплексообразования. Рассмотрим систему:
Должна идти следующая реакция: Co3+ + NO → Co2+ + NO2- На самом деле идет реакция: (CH3COO)2 +7KNO2 +2CH3COOH = 4CH3COOK +K3[Co(NO2)6] +NO+H2O
ЛЕКЦИЯ №7
|
||||||||||||||||||||
|
|
Последнее изменение этой страницы: 2017-01-25; просмотров: 2529; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 216.73.217.86 (0.013 с.) |



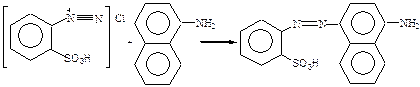



 ,
, 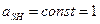



 - константа кислотности (1)
- константа кислотности (1) - константа основности (2)
- константа основности (2)

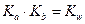
 , которое практически не влияет на pH.
, которое практически не влияет на pH.
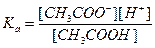













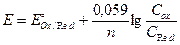
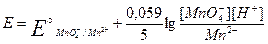
 и
и
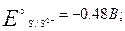 в кислой среде
в кислой среде